
Study Helps to Explain Symptoms Seen in Autosomal Dominant Form of Alport Syndrome
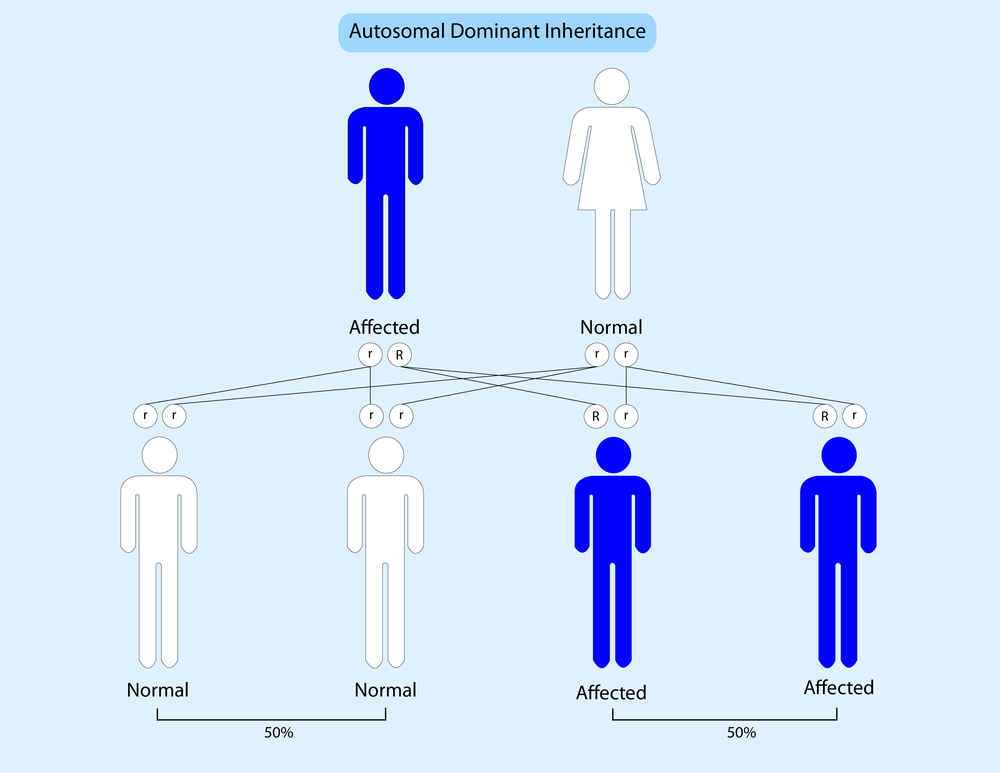



The inheritance patterns of Alport syndrome relate to the symptoms experienced by patients, researchers found. Symptoms associated with kidney function are much milder in autosomal dominant Alport syndrome than in forms of the disease with an autosomal or X–linked inheritance pattern, according to a study published in the Clinical Journal of the American Society of Nephrology.
“It may thus be difficult to make an accurate diagnosis of autosomal dominant Alport syndrome on the basis of clinical or pathologic findings,” the authors of the study, “Genetic, Clinical, and Pathologic Backgrounds of Patients with Autosomal Dominant Alport Syndrome,” wrote.
Researchers, led by Dr. Kazumoto Iijima with the Department of Pediatrics at Kobe University Graduate School of Medicine in Japan, analyzed 25 patients with genetically proven autosomal dominant Alport syndrome from 16 unrelated families, as well as their family members (a total of 72 people).
Patients’ median age when protein was first detected in the urine (a sign of kidney disease) was 17. One patient had hearing loss and another had eye lesions. A total of 16 patients underwent a kidney biopsy, and three showed focal segmental glomerulosclerosis (FSGS), a disease where scar tissue develops in the glomeruli or parts of the kidneys that filter the blood. In addition, seven patients who underwent kidney biopsy showed thinning of the glomerular basement membrane.
When the researchers analysed the patients genetically, they detected 13 different mutations causing the condition. Five of these were mutations that were seen to cause the autosomal recessive form of the condition in previous studies. Two of the families analyzed had mutations in two different genes involved in Alport syndrome.
Researchers concluded that patients with autosomal dominant Alport syndrome show a vast array of different symptoms, making it difficult to diagnose the condition, especially in the early stages. This can lead to some patients being diagnosed incorrectly.
In most patients with Alport syndrome, the condition is inherited in an X-linked pattern. The condition can also be inherited in an autosomal recessive manner, where the disease only develops when two copies of the faulty gene are inherited (one from each parent). The clinical, genetic, and pathologic backgrounds of patients with the autosomal dominant form of the condition, where only one faulty copy of a gene is enough to cause disease, remain unclear.
This study provides some clarifications to this background in a relatively large number of patients.



Leave a comment
Fill in the required fields to post. Your email address will not be published.