
Case Report: Tissue Biopsy Essential to Diagnose Complex Cases of Kidney Disease
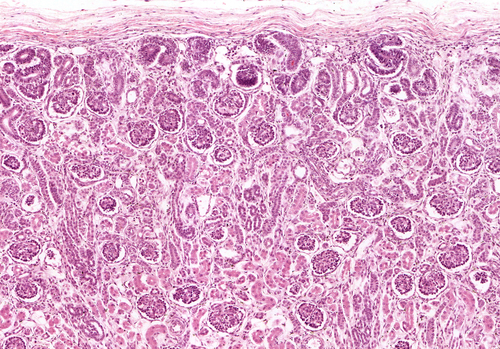



Analysis of kidney tissue can be an essential step for accurate diagnosis of complex conditions affecting the filtration function of the kidneys, contends a case report published in the journal Transplantation Proceedings.
Alport’s Syndrome is a hereditary disease caused by genetic defects in the gene that encodes the several chains that form the type IV collagen, the major structural component of glomerular basement membranes; it is an essential component of the kidney’s filtration system.
In the study “Thin Glomerular Basement Membrane in a Kidney Transplant of an Alport’s Syndrome Patient: A Case Report” a research team at Vanderbilt University Medical Center presented the rare case of an Alport’s syndrome patient who developed thin glomerular basement membrane upon renal transplantation.
At initial evaluation, the patient, a 20-year-old Indian man, presented several signs of Alport syndrome. He had increased blood pressure (hypertension), traces of blood in the urine, increased levels of protein and creatinine in the urine, and bilateral swelling of the lower extremities. In addition, the patient had a family history of renal disease and kidney transplantation.
Additional blood analysis confirmed the symptoms were not due to an autoimmune disorder. Analysis of a kidney tissue sample revealed the patient had tissue fibrosis – tissue scaring with collagen deposition – and had a glomerular basement membrane thinner than normal. Evaluation of collagen fibers confirmed the diagnosis of X-linked Alport’s syndrome with severe, end-stage kidney disease.
The clinical team decided the best therapeutic option for this patient was kidney transplant, which he received from an unrelated living donor.
The donor had no history of kidney disease, although it was reported later he had “some hearing difficulties” and a problem with the lens in one eye, which can be symptoms of Alport syndrome, the researchers stated.
Five months after the surgery the patient developed acute kidney injury, with increased blood creatinine levels, and increased urine protein and creatinine levels, as well as persistent blood in the urine.
Analysis of a tissue sample collected from the transplanted kidney revealed no alterations in collagen IV fibers. However, mild fibrosis lesions and glomerular basement membrane thinning were detected. Based on these symptoms and clinical evidence, the team reached a diagnosis of carrier state of Alport syndrome for both the kidney donor and now the patient.
“This case demonstrates the value of kidney biopsy in assessing patients with complex glomerular conditions, revealing a double disease state,” the authors stated.
The patient was treated with 500 mg of methylprednisolone for three days, which allowed the transplanted kidney to recover. The patients initiated immunosuppression therapy with 360 mg of Myfortic (mycophenolic acid) twice a day, Prograf (tacrolimus) 2 mg twice a day, and 5 mg of prednisolone daily. The patient remained stable with no report of disease progression until June 2016.
“Female carriers of X-linked AS typically have a good renal outcome, but about 15% develop ESKD [end-stage kidney disease] by the age of 60 years. Thus, the carrier state should be viewed as an at-risk rather than a benign condition,” the researchers wrote.
“Carrier female family members of those AS patients who have progression to ESKD should be strongly discouraged from donating a kidney,” they added.


Comments